
INTRODUCCIÓN
El cáncer de ovario (CO) es una neoplasia maligna que ocupa el 18vo lugar por frecuencia de cáncer y 14vo por decesos por cáncer en mujeres a nivel mundial [1,2]. En México, representa la tercera causa de muerte por tumor maligno en mujeres de entre 30 a 59 años [3].
Los factores de riesgo asociados al CO son los siguientes:
1) la edad, siendo el promedio de presentación entre 60 y 65 años, de los cuales el 75% se manifiestan posterior a la menopausia; sin embargo, aquellas mujeres que portan variantes en los genes BRCA1/2 pueden desarrollar la enfermedad a una edad más temprana.
2) La genética, donde las variantes en estos genes BRCA implica que el riesgo acumulado promedio de CO a los 70 años sea del 59%, e involucra la asociación con síndromes de susceptibilidad a cáncer, como es el síndrome de cáncer de mama y ovario hereditarios (SCMOH), el síndrome de Lynch, el Síndrome de Li Fraumeni, entre otros.
3) La existencia de antecedentes familiares de CO y otras neoplasias asociadas, como cáncer de mama y próstata, incrementan el riesgo entre 3 y 4 veces.
4) Historial de cáncer previo, por ejemplo, las mujeres que han padecido cáncer de mama con receptores de estrógenos negativos y que además han tenido exposición a radiación, presentan un 24% de riesgo a desarrollar CO en comparación con la población general.
5) Menopausia tardía, que se define como la ocurrencia de este evento a los 55 años o más, lo cual, se encuentra correlacionado con el número total de ciclos menstruales experimentados a lo largo de la vida de una mujer.
6) Nuliparidad, que se asocia a un incremento del 24% en el riesgo en comparación con aquellas mujeres que han dado a luz al menos a un hijo, y este riesgo se aumenta hasta 12 veces al compararlas con mujeres multíparas.
7) Tabaquismo, donde fumar de forma activa y regular genera un riesgo del 85% al 125% en personas que desarrollan CO del tipo mucinoso, en comparación con mujeres que no fuman.
8) Dietas ricas en grasas, especialmente comunes en países de América del Norte y Europa occidental, donde prevalece el consumo de grasas saturadas de origen animal y una menor ingesta de fibra.
9) Haber sido sometida a procedimientos in vitro, donde se describe un aumento de riesgo cercano a 20 veces. Por último, el estrés psicológico, también se reconoce como un factor de riesgo relevante para el CO, debido a que las modificaciones en los estilos de vida fomentan conductas de alto riesgo, tales como dietas desequilibradas, sedentarismo y consumo de sustancias [4 – 6].
En cuanto a los síntomas del CO estos pueden ser imprecisos al inicio de la enfermedad, manifestándose como dolor tipo cólico en el flanco pélvico afectado y/o distención abdominal, además de urgencia urinaria. En las etapas más avanzadas los síntomas pueden incluir dificultad respiratoria, secundario a la presencia de ascitis o derrame pleural, así como la presencia de un tumor palpable o visible por ultrasonograma o tomografía [7].
La clasificación histológica del CO según su estirpe celular y grado de malignidad puede ser de tres tipos: tipo 1 de células epiteliales, tipo 2 de células germinales y tipo 3 de células del estroma [6, 8]. El CO tipo 1 de células epiteliales es el más frecuente, ya que se presenta en aproximadamente el 90% de los casos, su incidencia aumenta con la edad, mostrando su pico más alto durante la perimenopausia [1, 4]. Este a su vez, se divide en varios subtipos epiteliales, como son: seroso de alto grado con una frecuencia del 70-80%, endometroide con frecuencia de 10-20%, de células claras y seroso de bajo grado, ambos con una frecuencia de 5-10%, mucinoso del 3-5% y carcinosarcoma del 1-3% [1, 8].
El carcinoma seroso de alto grado (CSAG), además de ser reconocido como el tipo más prevalente y grave, también se considera el responsable de aproximadamente el 70 al 80% de la mortalidad asociada con CO [7]; esto se debe a la presencia de múltiples factores genéticos, como sería la presencia de variantes que alteran la función proteica de TP53, el cual, podría ocurrir hasta en el 96% o la presencia de variantes en genes BRCA, que se pueden observar en hasta un 20% de las mujeres con CO de origen hereditario [7, 9]. Identificar a mujeres con variantes en los genes BRCA1 y BRCA2 repercute en las tasas de supervivencia general, ya que existen tratamientos específicos, como son los inhibidores de la PARP (poli(ADP-ribosa) polimerasa) [7], una terapia sintética que elimina las células con defectos en los mecanismos de reparación del DNA de doble cadena, provocados por la presencia de variantes en estos genes, contribuyendo así a la eliminación dirigida de las células [10].
Según Pietragalla A., et. al., el cáncer de ovario, además, podría ser parte de un síndrome de cáncer hereditario en un 23% de los casos y debido a su asociación con variantes en genes BRCA (la mayoría de los que producen una proteína trunca), también podría estar asociado con otros tipos de cáncer como serían, páncreas, próstata, laringe, estómago, colon y melanoma, entre otros [9]. Las pacientes portadoras de una variante en el gen BRCA1, tendrán un riesgo de padecer cáncer de ovario del 15-45% y las portadoras de variantes en BRCA2 el riesgo será del 10 al 20%. Sin embargo, entre el 10-15% de pacientes con cáncer de ovario seroso de alto grado no presentarán una variante deletérea, sino más bien, presentarán variantes de significado incierto (VUS) que deben ser analizadas y reclasificadas, tomando en cuenta la historia familiar y antecedentes personales de las pacientes [4, 11].
Por su localización, su velocidad de crecimiento, su sintomatología inespecífica, escasos factores de riesgos a los que se asocia y la falta de una prueba de tamizaje, condicionan que la detección del CO sea generalmente en estadios clínicos avanzados y que sólo el 15% de los casos estarán confinados al ovario al momento del diagnóstico [4]; por tal motivo se le considera uno de los cánceres ginecológicos más letales y, en consecuencia, un importante problema de Salud Pública.
Identificar, diagnosticar y clasificar a las pacientes con CO, se vuelve un reto para el médico Genetista y Oncólogo sin la asistencia de una genealogía o un estudio molecular; ya que, existen casos únicos en la familia que por edad de presentación nos podrían guiar hacia un síndrome de cáncer hereditario o genes accionables que podrían modificar el manejo integral de las pacientes. Por lo que, el objetivo de este trabajo es describir desde una perspectiva genética y molecular, las características de una muestra de pacientes con carcinoma de ovario seroso de alto grado (CO-SAG), con el fin de apoyar la toma de decisiones médicas y promover un abordaje preciso y efectivo para las pacientes con este tipo de tumor.
MATERIALES Y MÉTODOS
Se realizó un estudio prospectivo, observacional, abierto, descriptivo y transversal, durante el periodo de 2017 a 2019, aceptado por los Comités locales. Se incluyeron mujeres mexicanas referidas de la consulta de Oncología Médica, con diagnóstico de cáncer de ovario epitelial de tipo seroso de alto grado (CO-SAG) confirmado por estudio histopatológico. Las mujeres seleccionadas fueron enviadas a la consulta de Genética Oncológica para ser evaluadas por un oncogenetista; todas las mujeres firmaron la carta de consentimiento informado, previo asesoramiento genético, sobre el significado del posible resultado del estudio molecular de los genes BRCA1 y BRCA2. Posteriormente, a cada una de ellas se les solicitaron datos epidemiológicos y clínicos de interés, que incluían, lugar de residencia, edad, edad de diagnóstico, antecedentes familiares de cánceres, estadio clínico de la enfermedad, entre otros.
A cada una de las mujeres se le realizó una genealogía para investigar si el CO podría ser parte de algún síndrome de susceptibilidad a cáncer como el Síndrome de Cáncer de Mama y Ovario Hereditario (SCMOH), el Síndrome de Li-Fraumeni o el Síndrome de Lynch, según los criterios establecidos por National Cancer Institute [12] o si era un CO de tipo familiar (antecedentes familiares con varios tipos de cáncer, sin la presencia de variantes germinales) o esporádico (sin antecedentes familiares de cáncer).
El estudio molecular de los genes BRCA1 y BRCA2 se realizó en un laboratorio externo a nuestra Institución, siendo el análisis molecular por secuenciación de última generación (NGS, por sus siglas en inglés) y/o por estudio de grandes rearreglos, en caso de no tener resultados por NGS. Se recibieron los resultados vía correo electrónico y se citaron a las pacientes a la consulta de oncogenética para completar el asesoramiento genético.
Los antecedentes epidemiológicos de cada paciente se registraron en una base de datos construida en un software de hoja de cálculo para organizar, analizar y visualizar los datos para su análisis posterior.
RESULTADOS
Se incluyeron un total de 13 mujeres de origen mexicano, con diagnóstico de CO-SAG, que tenían una edad promedio de 50±10.25 años, de las cuales, el 76.9% era residentes de la Ciudad de México y el 38.4% eran solteras. En los antecedentes gineco-obstétricos se observó que el promedio de la menarca en las mujeres fue de 11.9 años, todas las mujeres presentaron un ritmo regular; el 26.3% no tuvieron gestas mientras que el 47.4% el nacimiento de sus hijos fue mediante parto (P), 15.8% fue por cesárea (C) y 10.5% tuvo un aborto (A) (Tabla 1).
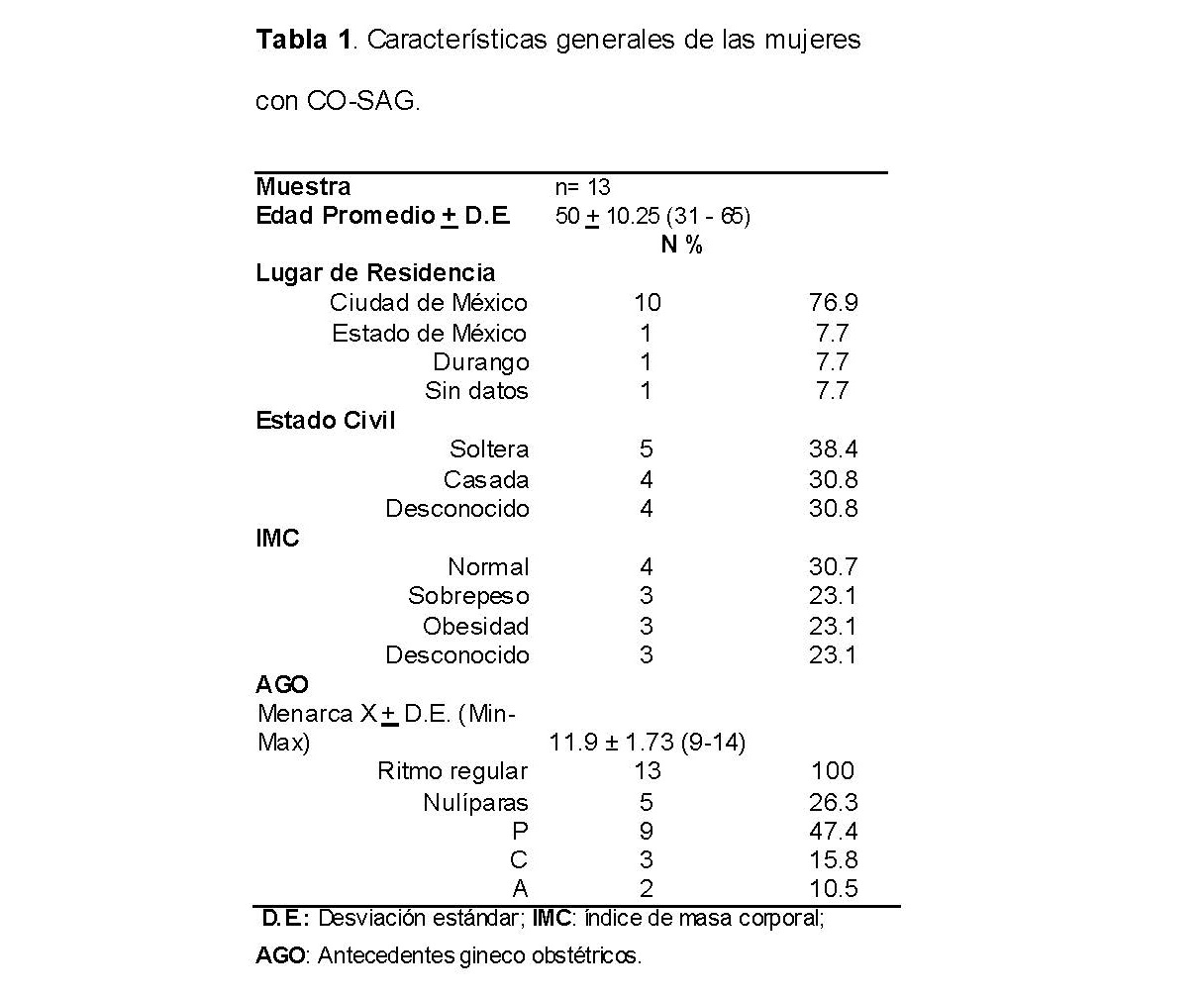
En relación a los antecedentes heredofamiliares de cáncer (AHF), el 92.3% (n= 12) de las mujeres presentaron antecedentes positivos, donde los tipos de cáncer más frecuentemente detectados fueron el cáncer de mama (17.14%), seguido de cáncer de ovario (14.28%) y de colon (8.57%) (Ver Figura 1).
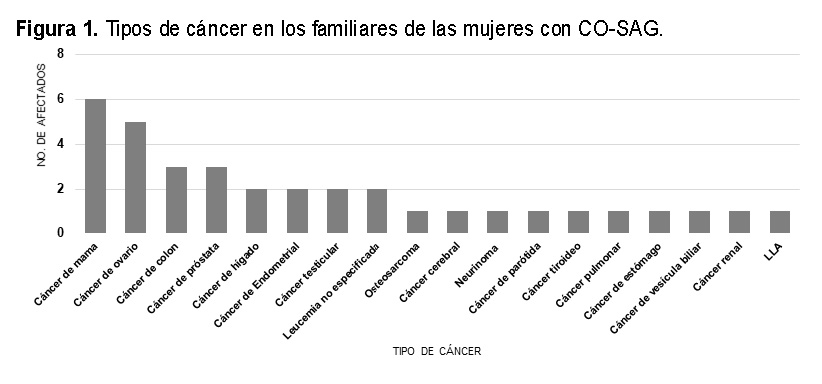
La edad de diagnóstico promedio en las mujeres estudiadas fue de 47.85 ± 11.25 con un rango de 31 - 65 años; al agruparlas por decenios observamos que más del 70% de ellas, se diagnosticaron entre los 30 y 50 años (Ver Figura 2A). Características clínicas e histopatológicas generales en las mujeres con CO SAG. A) Edad de diagnóstico.
Además, la mayoría de las mujeres presentaron síntomas inespecíficos asociados a colitis o dolor abdominal tipo cólico, entre los más frecuentes, (Ver Figura 2B).
En cuanto a la estadificación del CO, se encontró que el estadio clínico predominante en las mujeres con CO SAG fue el estadio IV, en un 69.2% de los casos, seguido por el estadio III y estadio I, en un 23.1% y 7.7%, respectivamente (Figura 2C).
Al momento del diagnóstico, la lateralidad predominante del tumor ovárico en las mujeres estudiadas fue el unilateral en un 61.5% (n = 8), solo el 7.7% (n = 1) de los casos se presentaron con tumor bilateral y el 30.8% (n = 4) no se encontraron datos en el expediente de la lateralidad del tumor (Ver Figura 2D).
El diagnóstico con el cual fueron referidas todas las mujeres por parte del Servicio de Oncología al servicio de Oncogenética fue probable síndrome de susceptibilidad hereditaria a cáncer, sin embargo cuando se realizaron las genealogías se pudieron clasificar en 5 tipos de Síndrome de cáncer hereditario, los cuáles incluyeron al síndrome de cáncer de mama y ovario hereditarios (SCMOH), síndrome de Li Fraumeni, síndrome de Lynch, probable cáncer asociado a mutaciones en CDH1 y por último, los casos dudosos, aquellos que, debido a que compartían varios tipos de cáncer entre ambas líneas parentales, no se les pudo clasificar en un tipo específico (Tabla 2).

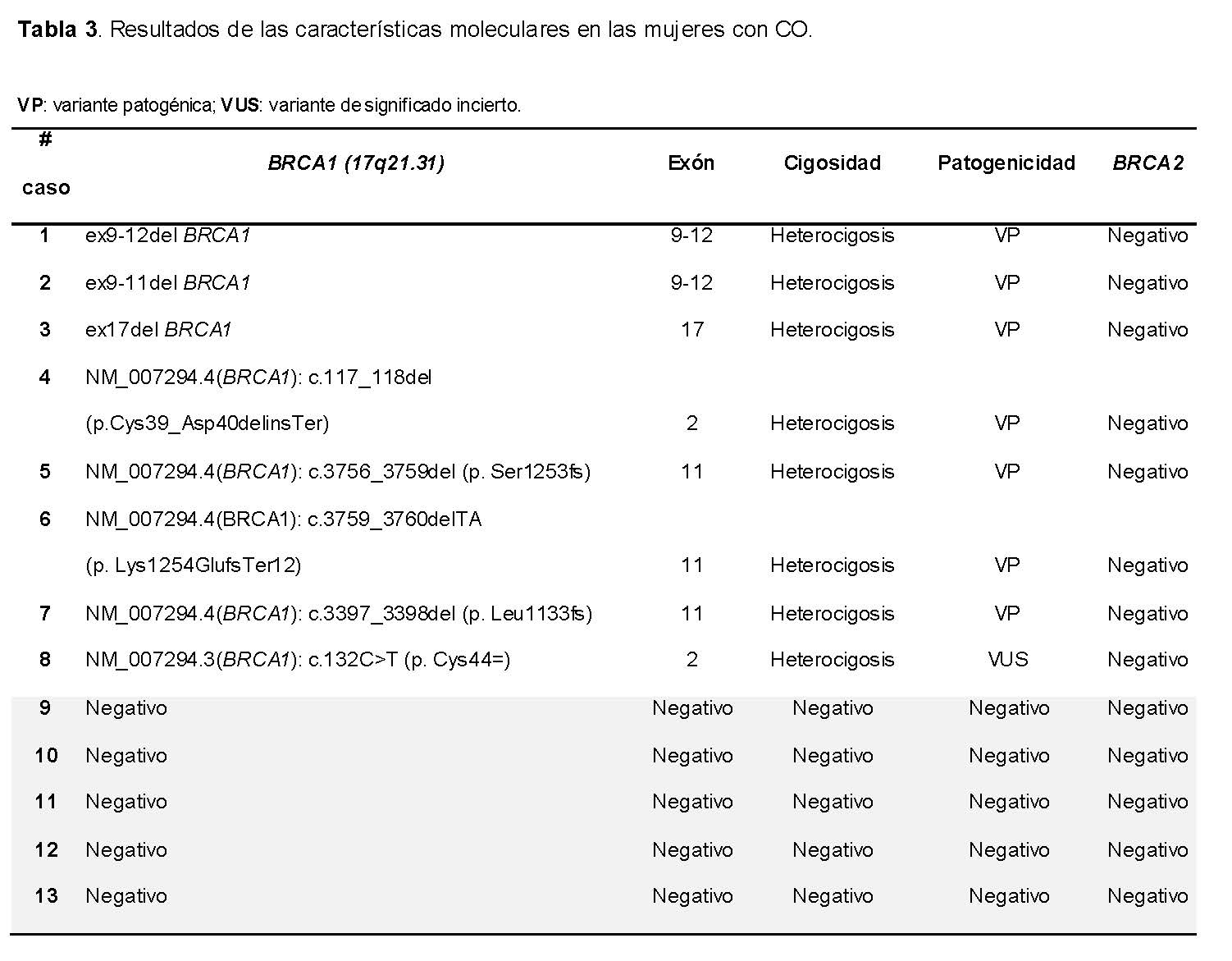
Los estudios moleculares de los genes BRCA1/2, mostraron que solo en algunos casos se encontraron variantes para el gen BRCA1, mientras que para el gen BRCA2 no se encontró ninguna variante.
En la Tabla 3 se muestran las características moleculares del gen BRCA1 en el locus 17q21.31 encontradas en el 61.5% (n= 8) de las mujeres con CO-estudiadas, de las cuales, el 15.4% (n= 2) se presentó la deleción de exones 9-12, considerada variante patogénica (VP) fundadora de la población mexicana; el 7.7% (n= 1) tuvo deleción del exón 17 considerada VP; el 15.4% (n= 2) mostró una variante intrónica en el sitio donante de empalme y una alteración de un nucleótido no conservado que implica cambio de marco de lectura en el exón 2, la primera considerada VP y la segunda probablemente patogénica (VPP); y el 23% (n= 3) presentaron variantes que produce un cambio de marco de lectura que implica la alteración de un nucleótido no conservado, consideradas VP. Cabe destacar que las 3 variantes caen en “ovarian cancer cluster región” (OCCR) en el exón 11 de BRCA1, una región que en el SCMOH preferentemente predispone a manifestarse como cáncer de ovario.
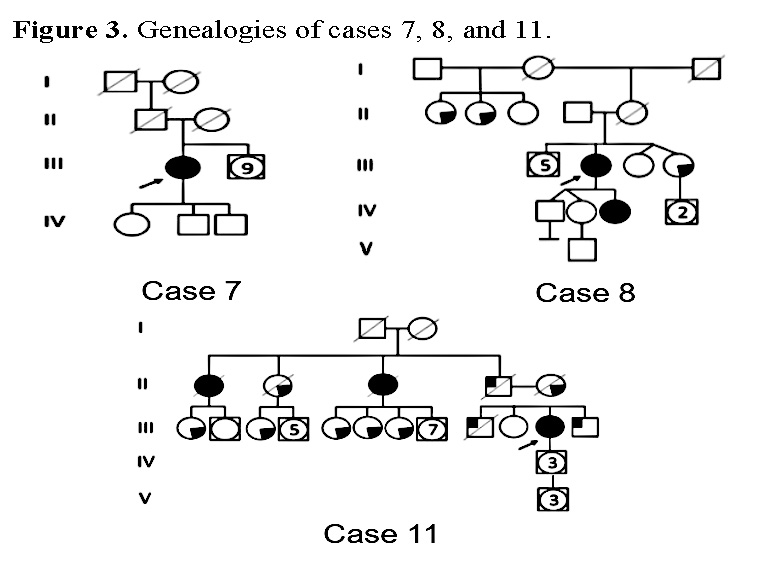
Con respecto a las genealogías representativas para un asesoramiento genético pudimos observar 3 casos específicos. El caso 7 que presenta una VP no tiene ningún antecedente de CO u otro tipo de cáncer en su genealogía y además es una paciente joven (38 años). El caso 8 con una VUS, muestra en su genealogía antecedentes de cáncer de ovario y otros tipos de cáncer relacionados en varias generaciones. Por otro lado, el caso 11 fue negativo para variantes tanto en los genes BRCA1 y BRCA2 y, sin embargo, muestra varios familiares con CO y otros tipos de cáncer en su genealogía (Figura 3).
Según el National Comprehensive Cancer Network (NCCN) [13], si los resultados de los genes BRCA fueron negativos, se deberán de considerar otros genes prevalentes asociados al CO, como sería el gen TP53 del Síndrome de Li-Fraumeni y los genes del sistema Mismatch Repair (MMR): MLH1, MSH2, MSH6, PMS2 o EPCAM del Síndrome de Lynch como la segundas y terceras opciones de estudio.
DISCUSIÓN
Los estudios epidemiológicos de CO muestran que para el año 2045, el aumento de casos será del 40.4%, con un consecuente acrecentamiento en la mortalidad en un 53.2%, además, los países con ingresos económicos bajos y medios como lo es México, tendrán un problema de salud pública por la falta de estrategias de detección, vigilancia, atención y tratamientos novedosos para esta patología [14].
Cuando se realiza el abordaje de las mujeres con CO, los antecedentes familiares, el análisis de la genealogía, los estudios histopatológicos, y de gabinete, nos ayudan a la toma de decisiones precisas para el reconocimiento de la enfermedad y para proporcionar un asesoramiento y seguimiento para el CO en mujeres en edad reproductiva y productiva.
Lamentablemente, el diagnóstico de la enfermedad se efectúa en estadios clínicos avanzados, debido a que, la sintomatología del cáncer de ovario es muy inespecífica al inicio de la enfermedad [6]. Lo cual se demostró en este estudio donde la mayoría de las mujeres con CO tuvieron al menos un año anterior, síntomas vagos manifestados como dolor abdominal tipo cólico o colitis y fueron diagnosticadas a una edad entre 30 - 50 años en estadios clínicos avanzados III y IV.
Por lo anterior, se vuelve imperante, la detección de CO a través de los 2 genes transcendentales asociados al desarrollo de este tipo de cáncer, ya que, las mujeres portadoras de variantes en genes BRCA1/2 y sus familiares, tienen un riesgo permanente de CO del 39%-44% si son portadoras de variantes en BRCA1 y del 11%-17% si son portadoras de variantes en BRCA2. Además de que estos genes son importantes para el reconocimiento de riesgos, evaluación predictiva y abordaje terapéutico en CO [6, 15].
Es fundamental resaltar la importancia del asesoramiento genético previo a la realización del estudio molecular de BRCA1/2, para determinar si las mujeres con CO desean proceder con el estudio, dado que se trata de una elección autónoma y voluntaria, que implica la necesidad de informar y dar soporte a la paciente sobre los posibles riesgos, el tipo de herencia en la familia y sus implicaciones médicas, con la finalidad de tomar decisiones fundamentadas en datos. De manera similar, el asesoramiento genético posprueba, sirve para identificar la causa principal, analizar de forma específica los resultados y proporcionar información relativa a la prevención, a las estrategias de manejo, así como a los posibles tratamientos que se encuentren disponibles [16, 17].
De esta manera, se evidencia a través de la genealogía y los resultados moleculares, que un considerable número de mujeres diagnosticadas con CO, tienen una historial familiar de este tipo de cáncer y otras neoplasias asociadas, lo que sugiere su inclusión en un síndrome de susceptibilidad a cáncer.
Entre los síndromes más comúnmente identificados se encuentran el síndrome de cáncer de mama y ovario hereditario, el síndrome de Li Fraumeni y el síndrome de Lynch. Sin embargo, en uno de los casos analizados (caso 6), se observa que no es imprescindible contar con antecedentes familiares de cáncer para suscribir la posibilidad de un síndrome de susceptibilidad a cáncer y, por ende, la portación de una variante germinal en BRCA1/2; la temprana aparición de la enfermedad podría ser el único indicativo para considerar la existencia del síndrome de susceptibilidad a cáncer.
Para las mujeres portadoras de variantes en genes BRCA1 se recomienda la cirugía de reducción de riesgo como la salpingooforectomía bilateral, si realizaron plenamente la maternidad y se encuentran entre los 35 y 40 años, o en mujeres entre los 45 y 50 años clasificadas dentro del síndrome de Lynch, ya que reduce el riesgo de CO en un 81% durante los 4 años siguientes a la resección [1,13]. Sin embargo, existe la posibilidad de padecer síntomas vasomotores, alteraciones en la función cardiaca, problemas cognitivos, osteoporosis, entre otros, si no se tiene las medidas preventivas adecuadas, debido a los efectos de una menopausia quirúrgica prematura [1,13].
Las mujeres en estadios iniciales de CO y que deseen tener hijos pueden optar por la salpingooforectomía unilateral, ya que las tasas de supervivencia a 5 años varían del 100% al 66%, dependiendo el subtipo de cáncer y estadio clínico, respectivamente. También se ha descrito que en las mujeres jóvenes portadoras de variantes en genes BRCA que no son sometidas a salpingooforectomía bilateral, se les puede tratar con anticonceptivos orales, los cuales reducen el riesgo de CO en un 36% [1,13].
Con los datos anteriormente citados, tenemos que para el caso 7 (Figura 3), el asesoramiento genético consistió en informar a la paciente de una posible mutación “de novo”, ya que no existe ningún otro familiar afectado con cáncer, así como el riesgo que tienen de heredar la variante, por lo que se sugieren estudios de extensión para su familia, ya que la variante está localizada en el OCCR y se recomienda terapia con inhibidores de PARP.
En el caso 8, que inicialmente se clasifico como una VUS, en el asesoramiento genético se le informó, que estaría en vigilancia, ya que con el tiempo la variante podría modificar su patogenicidad (como sucedió en este caso), que pasó por VPP a VP.
El caso 11 presenta varios familiares con CO y otros tipos de cáncer, sin embargo, presenta un estudio molecular negativo para variantes en BRCA1/2; se brinda asesoramiento genético para un síndrome de susceptibilidad hereditaria a cáncer y se propone extender el estudio molecular para la paciente y sus familiares.
Las normas establecidas de la Red Nacional Integral del Cáncer (NCCN) establecen realizar pruebas genéticas de otros genes de susceptibilidad para CO, cuando no se hayan tenido resultados positivos en BRCA1/2, como son PT53 en síndrome de Li-Fraumeni, PTEN en Síndrome de Cowden, STK11 en síndrome de Peutz Jeghers, MLH1, MSH2, MSH6 en síndrome de Lynch, además de ATM, PALB2, RAD51C y RAD51D, entre otros [1, 9,13].
CONCLUSIONES
Nosotros presentamos una casuística de CO SAG en donde la mayoría de los casos presentaron antecedentes familiares y genealogías compatibles con un síndrome de cáncer de mama y ovario hereditarios.
Los estudios histopatológicos y moleculares son de gran trascendencia en pacientes con CO SAG, ya que este tipo histológico de cáncer se asoció, en este estudio, con el 61.5% de los casos al gen BRCA1.
El 23% de las mujeres con CO SAG la variante se localizó en el exón 11 de BRCA1 en la región conocida como OCCR, una región que predispone a una mayor susceptibilidad para cáncer de ovario. Mientras que el 15.4% de las mujeres con CO SAG presentaron deleción de los exones 9-12 del gen BRCA1, descrita como mutación fundadora en población mexicana para SCMOH.
CONTRIBUCIÓN
MOQ: Conceptualización, Curación de datos, Análisis formal, Metodología, Administración del proyecto, Supervisión, Validación, Redacción (borrador original), Redacción (revisión y edición), LGO: Metodología, Validación, Redacción (borrador original), Redacción (revisión y edición). GCS: Redacción (borrador original), Redacción (revisión y edición).